

One of the organisms of choice for the production of recombinant proteins is Escherichia coli. It has become the most popular expression platform and its use as a cell factory has been well established. As a result of this, many molecular tools and protocols are at hand for the production of high-level heterologous proteins such as a vast catalog of expression plasmids, lots of engineered strains, and many cultivation strategies. E. coli is used for the production of recombinant protein because it serves as the simplest, fastest, and most cost-effective system for the custom expression of the protein. Production of recombinant proteins in E. coli is a preferred solution when the final application requires that large quantities of proteins such as crystallography and nuclear magnetic resonance (NMR) and no Post-Translational Modifications are required. Bacterium Escherichia coli is the most widely used host for producing recombinant proteins, and the majority of the recombinant proteins are produced in the cytoplasm of E. coli.
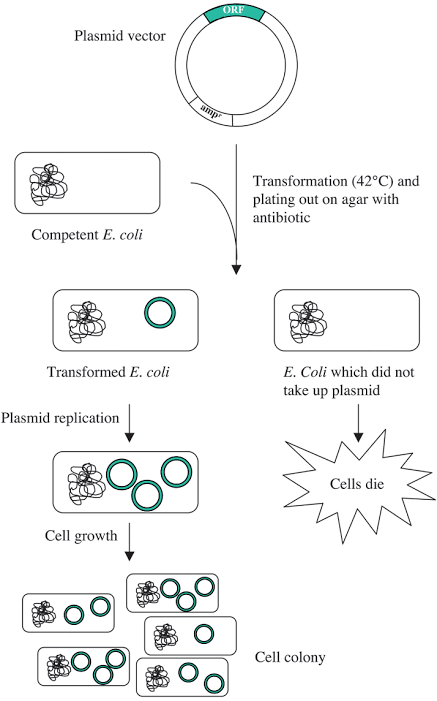
It is therefore common to produce recombinant proteins that contain disulfide bonds in the periplasm. By targeting the protein to the periplasm, it becomes possible to take advantage of the oxidizing environment and the resident disulfide bond formation system in facilitating accurate disulfide bonds. An expression system involved in the production of recombinant proteins in E. coli mostly entails the combination of a plasmid and a strain of E. coli. The main aim of recombinant protein expression service is often to obtain a high degree of accumulation of the soluble product in the bacteria cell. This strategy is not always accepted by the metabolic system of the host. In some situations as well, a cellular stress response can be encountered. In recombinant systems, another response that is always encountered is the accumulation of target proteins into soluble aggregates that are known as inclusion bodies. These aggregated proteins are typically misfolded which results in biological inactiveness.
Materials and methods for the recombinant protein production
Construction of E. coli W3110/RHA/lac

In deleting the RHA operon and the lac operon in W3110, the Red-swap-method was employed. In the use of pKD13 plasmid as a template and the primer pairs, kanamycin cassettes with regions homologous to the 5’ and 3’ flanking regions of the RHA operon and lac operon were generated with PCR. The template was further digested with Dpnl and the molecular weight of the PCR products was verified using an agarose gel and isolated using the Thermo Fisher Scientific gel extraction kit. In generating the W3110rha KmRand W3110lac KmRstrains, the purified PCR products were electroporated into W3110 cells harboring Pkd46 that had been cultured at 30oC in standard Lysogeny broth medium that contains 0.2%arabinrose. Thereafter, kanamycin-resistant clones (kan: 50ug/ml final concentration) were screened for proper kanamycin cassette insertion through PCR making use of primer pairs. In using P1-mediated generalized transduction, the region of interest of the strains becomes exposed to the lambda Red system and then transferred to cells that had not been exposed to the lambda Red system. When the transduction of the genetic region of interest became successful, cells were transformed with pCP20 to remove the kanamycin cassette from the genome through FLP-recombinase, and removal of the cassette became verified through PCR/sequencing. In the final stage, the cells became cured of pCP20 through protracted cultivation at 370C. For generating the W3110/RHA/lac strain which is the E. coli/RHA. The RHA operon in W3110 was deleted and the lac operon was also deleted from the resulting stain. The deletion of the lac operon prevented any secondary effects on the model recombinant proteinscFv BL1 that could occur as a result of binding to its substrate E. coli protein expression service.
Recombinant Protein Production Experiments

In the experiments of protein production, E. coli strain W310/RHA/lac was utilized. In this, cells were transferred with the expression of vectors or the empty expression vector that served as a control. Protein production screening was carried out in a standard 24-well plate format. Cells developed aerobically at 300C and 200 rpm (New Brunswick Innova 42R shaker with an orbit diameter of 1.9 cm), in LB medium that was supplemented with 50ug/ml kanamycin. For preculturing, 0.2% glucose was added to prevent the background expression of the genes encoding the targeted proteins. The growth was monitored through the measurement of A600 with a UV-1300 spectrophotometer. At the point of A600 of -0.5 targets gene expression was stimulated through the addition of rhamnose. The following rhamnose concentrations were used for scFv BL1 production screening: 0, 50, 100, 250, 500, and 5000 μM. While for the production screening of hGH, the following rhamnose concentrations were employed for use: 0, 10, 50, 100, 250, and 5000 μM. For further analysis, cells were harvested at specific points in time. In figures of culturing experiments, standard deviations were shown which were based on at least three independent biological replicates. Cultures for the isolation of hGH and BL1 were carried out in 2.5 L shake flasks (TunairTM) which contain 1 L of LB medium.
Conclusion
For the recombinant protein production purification using E. Coli, the protein expression service was easily gotten through a large test of different conditions. However, with how fast and simple e.coli makes the protein expression services, there are still some drawbacks to it which are the high amounts of endotoxins left in the final sample and the absence of Post-Translational Modifications. Although the removal of the endotoxins is feasible, it is not convenient at the industrial stage because there is not much that can be done in solving PTM problems other than the use of an expression system.
















{kind=link}